Building Regulatory Writing Capability in Small and Medium-Sized Companies
This article outlines how small and medium-sized medical device enterprises (SMEs) can build internal capacity for regulatory writing. It explores key regulatory writing modules, highlights tools such as QFD and risk management, and suggests resource strategies including outsourcing and knowledge capture.
QUALITY & REGULATORY
Mingyue Feng
5/13/202512 min read


Introduction
Medical Device Regulatory Writing (MDRW) is a demanding task that involves compiling structured, evidence-based documentation to demonstrate product safety, performance, and regulatory compliance. While large organizations often have established processes and dedicated teams to handle such documentation, small and medium-sized enterprises (SMEs) frequently face significant hurdles.
SMEs often lack sufficient internal resources, regulatory expertise, and systematized processes to meet increasingly complex regulatory requirements. This results in a fragmented understanding of MDRW—many teams can “write documents” but lack a clear framework of what to write, why, and for whom. Moreover, key personnel may hold tacit knowledge that is not institutionalized, and dependence on external experts often lacks strategic integration.
This article aims to demystify the core components of MDRW for SMEs by outlining essential topics, tools, and strategies. It proposes a structured roadmap that helps SMEs understand the why and how of MDRW: from clarifying design intent and clinical evidence, to ensuring traceable quality control, to managing documentation across the product lifecycle. Although this article focuses on medical devices, the underlying principles apply broadly across other regulated industries such as pharmaceuticals and cosmetics.
1. Defining Medical Device Regulatory Writing
Medical device regulatory writing (MDRW) refers to the specialized discipline of developing structured, scientifically grounded, and regulatory-compliant documentation to support the market authorization and lifecycle management of medical devices. MDRW plays a crucial role in the development of medical devices, ensuring that all documentation meets the stringent requirements set by regulatory authorities. This process integrates technical accuracy with clarity, ensuring alignment with the expectations of regulatory bodies such as the U.S. FDA, European Commission, and other national competent authorities.
Documents in scope typically include:
Clinical evaluation reports (CERs)
Technical documentation (including Design Dossiers and Device Master Records)
Risk management files (per ISO 14971)
Instructions for use (IFU) and labeling
Regulatory submissions (e.g., 510(k), PMA, CE marking applications)
Post-market surveillance (PMS) reports and periodic safety update reports (PSURs)
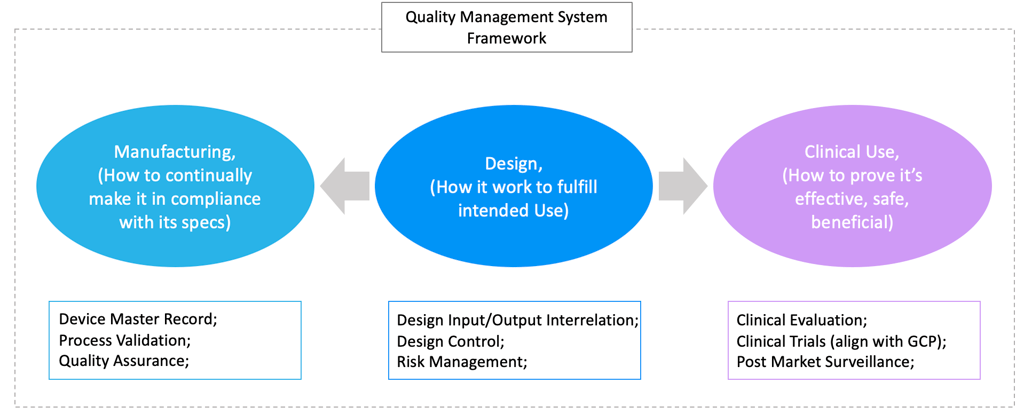
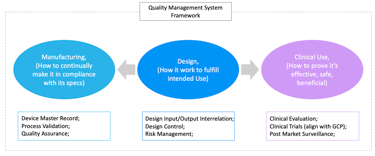
2. The Three Pillars of MDRW
A high-level understanding of medical device regulatory writing (MDRW) is not sufficient for regulatory success. Small and medium-sized enterprises (SMEs) must not only produce documentation that meets regulatory expectations, but also develop a structured understanding of the full range of regulatory writing responsibilities.
To make MDRW actionable and strategically valuable, it should be built around three foundational pillars that align with the product lifecycle and regulatory priorities:
Design: Ensuring Product Design Meets Intended Use
Clinical Use: Generating Evidence of Safety and Performance through Clinical Evaluation and PMS
Manufacturing: Establishing Process and Quality Controls to Ensure Product Conformity
These pillars are interconnected, bridging the design, clinical, and manufacturing stages of a product’s lifecycle. Together, they form a comprehensive framework for MDR documentation that withstands regulatory scrutiny.
Pillar 1: Design – Ensuring Product Design Meets Intended Use
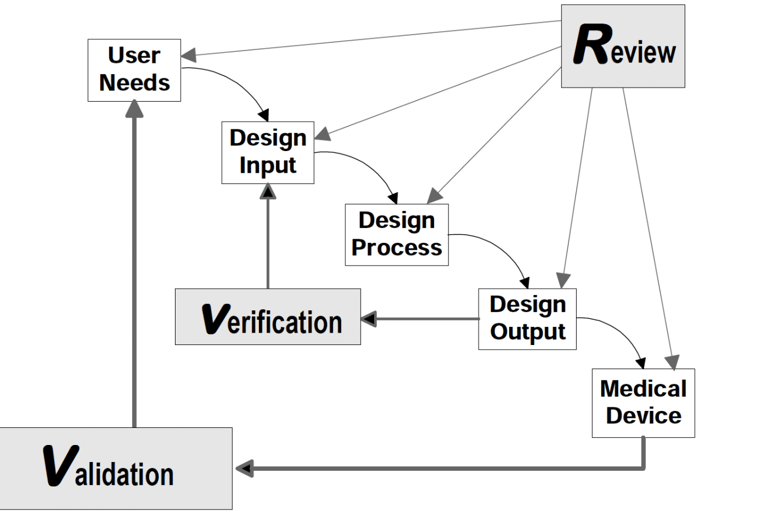
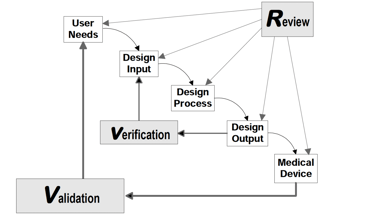
Understanding a medical device begins with a clear explanation of how it works and what it is intended to achieve. Simply listing components or final outcomes is insufficient. Regulatory authorities, particularly the FDA under 21 CFR Part 820, require traceable documentation that links user needs to design inputs, and design outputs to verified device features. The FDA’s Design Control Guidance for Medical Device Manufacturers provides a framework for implementing this traceability.
Two critical insights emerge from this model:
The ability to trace how each design input is translated into a design output—and subsequently into a physical product—is central to both compliance and product performance. Tools such as the Quality Function Deployment (QFD) matrix and the Design Input/Output Matrix provide structured mechanisms to document and visualize this traceability. These tools enhance team collaboration, support audit readiness, and ensure that complex user requirements are systematically addressed.
Design verification and validation serve as formal proof that the product meets its intended use. These activities must be conducted with scientific rigor and supported by objective, reproducible data. Verification confirms that outputs match input specifications, while validation ensures the device performs as intended in actual or simulated conditions of use.
Source: FDA Design Control Guidance for Medical Device Manufacturers
Pillar 2: Clinical Use – Demonstrating Safety and Performance in the Field
Clinical evaluation is a critical component of medical device regulatory writing (MDRW) and serves as a primary source of evidence for a product’s safety and performance. It is inherently multidisciplinary, drawing on clinical science, Good Clinical Practice (GCP), biostatistics, and systematic literature review.
A well-structured clinical evaluation typically includes the following elements:
Background and product description
Critical appraisal of existing clinical data (e.g., literature review)
Summary of clinical investigations, if applicable
Safety and performance analysis
Benefit–risk assessment
Conclusion on the sufficiency of clinical evidence to support market access or continued marketing
Regulatory authorities evaluate clinical documentation based on the validity of data, scientific integrity, ethical compliance, and the alignment between claims and evidence. For small and medium-sized enterprises (SMEs), which often lack dedicated clinical teams, engaging experienced third-party experts for protocol development, execution, and reporting can be both strategic and essential.
In addition to clinical evaluation, Post-Market Surveillance (PMS) plays a vital role in demonstrating continued safety and effectiveness after market approval. Regulatory bodies require manufacturers—including SMEs—to establish a PMS system that systematically monitors, collects, and analyzes real-world data throughout the device lifecycle.
A proactive and comprehensive PMS plan is more than a regulatory formality; it is a core component of risk management and lifecycle evidence generation. Authorities increasingly scrutinize how PMS data is used to update risk assessments, inform labeling, and support ongoing compliance with safety and performance requirements.
Pillar 3: Manufacturing – Establishing Capability to Ensure Product Conformity
MDRW must also address how a company ensures that its manufacturing processes consistently produce devices that conform to design specifications. This begins with the Device Master Record (DMR), which consolidates approved design outputs into a comprehensive specification set to guide production, inspection, and quality assurance.
To consistently meet the DMR requirements, the organization must implement a compliant Quality Management System (QMS) that integrates process controls, production planning, and supplier oversight. Regulatory focus typically centers on three key areas:
Process Validation – For processes where output cannot be fully verified through inspection (e.g., welding, sterilization), companies must demonstrate through validation protocols that the process parameters consistently yield compliant products. Validation methods and requirements are discussed in Chapter 3.
Quality Control – Key product attributes defined in the DMR must be monitored during production using statistically sound inspection and sampling plans.
Quality System Procedures – Documented procedures must be available and demonstrably implemented, covering areas such as production control, purchasing, receiving, and change management.
A mature QMS includes mechanisms for feedback from process monitoring and nonconformities, traceability between inputs and outputs, and evidence of continuous improvement. Auditors and regulators look closely at how these mechanisms ensure sustained product conformity throughout the lifecycle.
MDRW Operates Within the Quality Management System
Regulatory writing does not exist in isolation. Every MDRW activity should be integrated into the Quality Management System. The documents generated—whether related to design, risk, clinical evidence, or production—are part of the broader quality record that demonstrates compliance and organizational control. Embedding MDRW within the QMS ensures that documentation is traceable, auditable, and aligned with real-world operations.
3. Critical Techniques for Regulatory Writing
This chapter outlines five key techniques (Table 1) essential for effective Medical Device Regulatory Writing (MDRW). These methods support one or more components of MDRW, ranging from design development to post-market surveillance. They are often embedded within the Quality Management System (QMS) and typically require formal training for proficient use.
Technique 1: Quality Function Deployment (QFD)
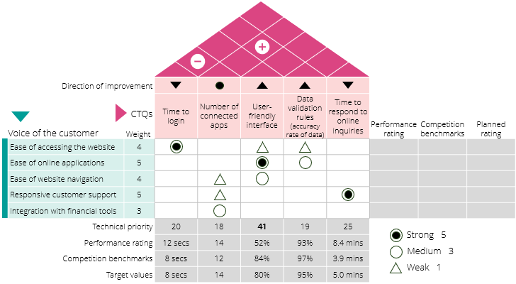
QFD is a structured, customer-centric approach used to convert customer needs into detailed product specifications. A central tool of QFD is the House of Quality (HoQ), a visual matrix that maps customer expectations against technical solutions. By structuring the design process around VoC (Voice of the Customer), QFD enhances clarity, collaboration, and prioritization. Key steps include identifying customer and technical requirements, building a relationship matrix, and setting design targets.
Quality Function Deployment (QFD) is a structured, customer-focused methodology used to translate customer needs into detailed product or service specifications, ensuring that the final product or service effectively meets those needs. It’s a systematic approach that helps organizations understand and prioritize customer requirements, and then design products or processes that deliver on those expectations.
A key tool in QFD is the House of Quality (HoQ), a graphical technique that Illustrates the relationship between customer requirements and product or process attributes. The diagram is named for its house-shaped appearance. It consists of multiple tables and matrices that analyze data sets according to the QFD objective. The House of Quality can play an important role in improving both the speed and quality of the QFD process, encouraging collaboration, and aiding in decision-making. Below is a typical example of House of Quality.
Technique 2: Design Input/Output Matrix (Design I/O Matrix)
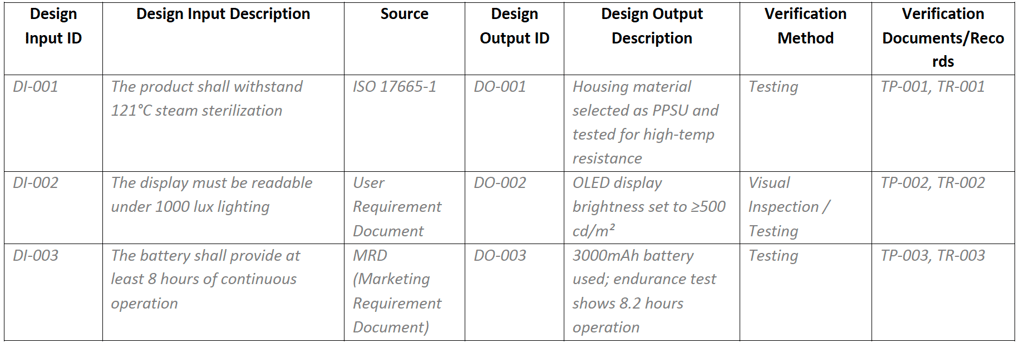
The Design I/O Matrix is another critical tool used in early design phases to ensure traceability between user needs (inputs) and design features (outputs). Unlike QFD, which focuses on requirement translation, the I/O Matrix ensures verification and validation. It supports alignment with Device Master Record (DMR) development and facilitates downstream quality control and auditing.
Below is an example commonly used across organizations, often integrated directly into the design output documentation. SMEs are encouraged to adopt and customize this format to suit their operational context.
Design Input/Output Matrix Example
Technique 3: Risk Management
Risk management is a core process under ISO 14971, supporting safety and effectiveness throughout the device lifecycle. Its scope includes:
Hazard Identification
Risk Evaluation (likelihood vs. severity)
Risk Control (design changes, protective measures, user warnings)
Benefit-Risk Analysis (when risk cannot be eliminated)
Residual Risk Acceptance
Post-market Monitoring
Two key characteristics are:
Dynamic: Risk files must evolve continuously with product changes.
Systemic: Risk control is integrated across the QMS—from design and supplier qualification to clinical feedback.
Common techniques include Failure Mode and Effects Analysis (FMEA) and Fault Tree Analysis (FTA), which help identify failure mechanisms early, particularly during development, design change, or post-market surveillance.
Technique 4: Process Validation
Process validation confirms that a manufacturing process can consistently produce products meeting specifications. Per GHTF SG3/N99-10:2004, validation is essential when:
The output cannot be fully inspected
Inspection would damage the product
Testing is inefficient or costly
A full validation report typically includes:
Scope and Product Description (IQ/OQ/PQ)
Definitions (e.g., critical process, revalidation)
Validation Standards (FDA QSR, ISO 13485)
Strategy and Risk Evaluation (e.g., FMEA)
Installation Qualification (equipment setup)
Operational Qualification (parameter checks)
Performance Qualification (batch tests under real conditions)
Sampling Plan and Criteria
Statistical Analysis (CpK, PpK, trends)
Deviation and CAPA handling
Revalidation Plan
Summary Report and Conclusion
Validation must be traceable, risk-based, and documented—key elements emphasized by regulators and auditors alike.
Technique 5: Statistical Techniques
Statistics play a central role in ensuring data-driven decision-making in MDRW. Regulatory reviewers frequently assess whether data sampling, analysis, and interpretation follow statistically sound principles.
Applications include:
Process Validation: DOE (Design of Experiments), regression
Process Monitoring: Control charts, Cp/Cpk, Pp/Ppk
Sampling Inspection: ANSI Z1.4, AQL systems
Clinical Trials: t-tests, chi-square, p-values, confidence intervals, effect sizes
Organizations should ensure that staff applying statistical tools have proper training and access to validated methods.
4: Resources Deployment
Small and medium-sized enterprises (SMEs) face unique challenges in building regulatory writing (RW) capability due to limited resources, diverse workloads, and often informal knowledge systems. This chapter explores how SMEs can leverage internal expertise, document templates, training programs, and external support to establish sustainable and scalable MDRW capacity.
4.1 Identifying Subject Matter Experts (SMEs)
Effective MDRW requires cross-functional input—no single person or department can manage the entire workload. Management should identify process owners to lead specific regulatory tasks, supported by relevant team members.
Each regulatory document (e.g., clinical evaluation, risk file, or DMR) should have a clear owner, ideally someone already responsible for the underlying process (e.g., R&D, QA, Clinical Affairs). As companies scale, SMEs may evolve from individuals to entire teams, each accountable for delivering compliant outputs within their domain.
4.2 Templates as Strategic Assets
While SMEs provide domain expertise, variability in knowledge and communication skills can compromise consistency. Therefore, well-designed templates are critical tools for regulatory alignment and quality assurance.
Templates should:
Embed structure and instructional guidance
Reference relevant standards and regulatory expectations
Be field-tested and updated based on feedback
Organizations can leverage templates brought in by employees from mature companies—but must ensure they are tailored to relevant regulatory contexts and not blindly reused. Overly complex templates intended for multinational submissions may be excessive for startups or regional MDR filings.
4.3 Training and Knowledge Sharing
Training is often undervalued in SMEs, where multitasking is the norm and dedicated time for learning can be scarce. Nonetheless, fostering a culture of structured knowledge sharing—both within and across departments—is essential to mitigate compliance risks.
Key practices include:
Internal training sessions to harmonize understanding of RW processes
Cross-project debriefings to share lessons learned
Regular reviews of regulatory updates and authority feedback
This requires leadership support and process incentives to prioritize learning alongside delivery.
Building Knowledge Management (KM)
Knowledge in SMEs is often trapped in people’s heads rather than documented processes.3 Early-stage companies are especially well-positioned to develop KM practices while teams are still small and communication is fluid.
KM involves:
Capturing knowledge from past projects and regulatory submissions
Structuring and storing documents in shared platforms (e.g., SharePoint, Notion)
Maintaining living documents that reflect ongoing regulatory changes
Enabling access through clear naming, version control, and indexing
This turns personal insight into institutional memory and strengthens business continuity.
4.4 Leveraging External Resources
While earlier sections focused on building internal capabilities, the scope of medical device regulatory affairs and quality system management is broad and complex. For small and medium-sized enterprises (SMEs), relying solely on internal resources is rarely sufficient. Strategic engagement of external resources is not just helpful—it is often critical for achieving compliance and accelerating development.
4.4.1 Engaging External Experts
One of the most impactful decisions SMEs can make is determining when, whom, and how to engage external expertise. This requires careful planning to identify the type of support needed, clarify expectations for deliverables, and ensure that the knowledge gained is systematically captured and reused within the organization.
As discussed throughout this article, MDRW spans three foundational pillars:
Product design and intended use
Clinical evaluation and demonstration of safety and performance
Quality system processes that ensure conformity
Among these, clinical evaluation is typically the area where SMEs require the most external support. Clinical evaluation reports (CERs), clinical investigation planning, statistical analysis, and benefit-risk assessments are highly specialized and often extend beyond the in-house capabilities of small teams.
To ensure value from external collaboration, SMEs should balance trust in the expert’s domain knowledge with active engagement in the development process. Reviewing protocol assumptions, literature sources, study designs, and analytical methods helps align documentation with the company’s internal understanding and prepares the team for potential regulatory questions. Collaborative critique improves both the quality of the deliverables and the organization’s preparedness for audits or technical file reviews.
Tip: A common pitfall for SMEs is treating a consultant’s report as the endpoint. In fact, high-quality deliverables and expert discussions represent valuable learning opportunities. These should be converted into internal knowledge assets—training materials, checklists, or editable templates—through knowledge management practices. Documenting lessons learned and standardizing successful approaches supports cross-functional learning and accelerates future regulatory activities.
4.4.2 Utilizing Regulatory Authority Resources
SMEs often perceive regulatory agencies solely as gatekeepers of approval. In reality, many agencies also serve as proactive educators, offering free and high-quality resources to support compliance and industry capability building.
Key opportunities include:
Accessing official guidance documents, webinars, and training portals maintained by regulatory bodies.
Submitting pre-submission dossiers to seek early-stage scientific or regulatory feedback, particularly for novel technologies or borderline classification cases.
Requesting live meetings or teleconferences (where available), which often offer richer engagement than written exchanges alone and allow for clarification of critical regulatory expectations.
These resources are not only valuable for new product development, but also for team training, SOP updates, and aligning MDRW practices with current expectations.
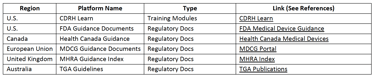
Table below provides examples of key platforms where SMEs can access training materials, regulatory interpretations, and submission-related guidance.
Regulatory Authority Learning/Sharing Platforms
Conclusion
MDRW is fundamentally the process of clearly articulating how a product meets requirements for safety, performance, quality control, and regulatory compliance. For small and medium-sized medical device companies, understanding the true “subject” of regulatory writing—that is, the diverse types of compliance documentation submitted to regulatory bodies—is the first step toward building effective regulatory capabilities.
This article aims to clarify the scope of MDRW by outlining the different types of documents and their respective regulatory objectives. For example:
Technical documentation must demonstrate a logical and closed-loop design process that integrates risk control.
Clinical evaluation reports must present credible evidence of clinical safety and performance.
Quality system documents must show systemic risk management and continuous improvement mechanisms.
Regulatory writing is not “writing” in the traditional marketing or editorial sense—it is the structured organization and presentation of data and logic in a way that aligns with regulatory expectations and withstands external scrutiny.
Our goal is to help SMEs adopt a clearer mindset toward MDRW: the objective is not merely to “submit paperwork,” but to build a transparent, auditable, and verifiable chain of regulatory evidence. Companies must learn not only what regulators need to see, but also how they expect to see it. By aligning documentation practices with regulatory logic, SMEs can avoid the common trap of producing content that is verbose yet uninformative—ultimately improving the efficiency of regulatory submissions and increasing the likelihood of approval.
References
FDA Design Control Guidance for Medical Device Manufacturers. https://www.fda.gov/media/116573/download
GHTF SG3/N99-10:2004 guidance document. https://www.imdrf.org/sites/default/files/docs/ghtf/final/sg3/technical-docs/ghtf-sg3-n99-10-2004-qms-process-guidance-04010.pdf
The critical success factors’ investigation during knowledge management implementation within SME enterprises: a Participatory Design opportunity. https://doi.org/10.1007/s42979-022-01420-6
The Ultimate Guide To Design Controls For Medical Device Companies. https://www.greenlight.guru/blog/design-controls?utm_source=chatgpt.com
The Ultimate Guide to Medical Writing for Medical Devices. https://www.rxcomms.com/learning/the-ultimate-guide-to-medical-writing-for-medical-devices?utm_source=chatgpt.com
21 CFR Part 50. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-50
S. FDA CDRH Learn. https://www.fda.gov/training-and-continuing-education/cdrh-learn
Health Canada Guidance. https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-devices/application-information/guidance-documents.html
MDCG Guidance Documents. https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en
MHRA Guidance Index. https://www.gov.uk/government/collections/regulatory-guidance-for-medical-devices
TGA Guidelines. https://www.tga.gov.au/standards-guidelines-publications-medical-devices-ivds
Quality Function Deployment: The Customer-Driven Methodology. https://www.6sigma.us/six-sigma-in-focus/quality-function-deployment-qfd/
Translating Customer Needs With Quality Function Deployment. https://citoolkit.com/articles/quality-function-deployment/
Related Reading:
ISO 14971:2019 Medical devices — Application of risk management to medical devices
Global Post-Market Surveillance (PMS) Regulation Highlights
Published on: May. 13, 2025