Beyond Compliance: Leveraging CAPA for Actual Improvement
CAPA is more than a compliance checkbox—it’s a key driver of quality and risk reduction in medical devices. This article explores CAPA’s regulatory role, common pitfalls, and how to turn it into a proactive tool for continuous improvement.
QUALITY & REGULATORY
Mingyue Feng
3/18/20254 min read


Introduction
If you work in the medical device industry, you’ve probably dealt with CAPA—Corrective and Preventive Actions.
Maybe you’ve written CAPA reports, responded to audit findings, or even sat through long meetings about root cause analysis. But let’s be honest: for many companies, CAPA is still treated as just another compliance checkbox rather than a real driver of quality improvement.
I’ve seen too many organizations scramble to close CAPAs before an audit, only to deal with the same problems resurfacing months later. Why? Because they’re focusing on fixing symptoms instead of solving root causes.
In this post, I want to shift the conversation. Instead of viewing CAPA as a regulatory burden, let’s look at how it can actually improve product quality, reduce risks, and drive business efficiency.
If you’re in Quality Assurance, Regulatory Affairs, or Operations, this post is for you. Let’s dive in.
1. CAPA and Regulatory Expectations: Why It’s a Big Deal
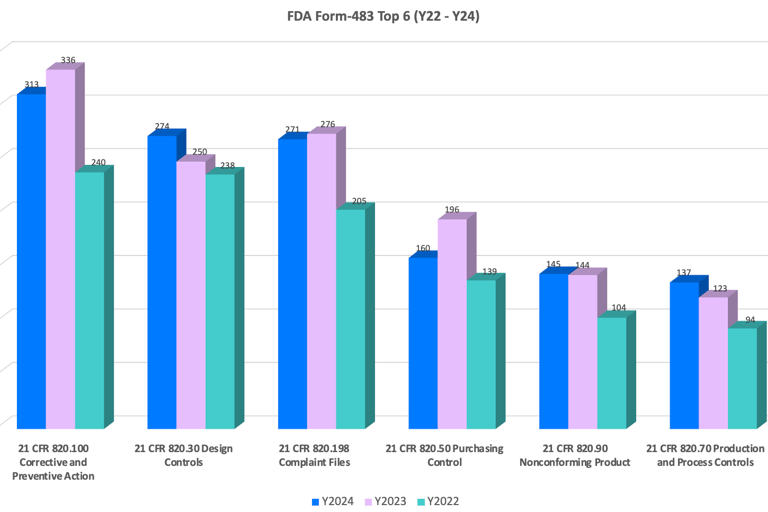
1.1 CAPA - One of the Most Common 483 Findings
Regulators across the world take CAPA very seriously. If you don’t manage CAPA well, you’re risking noncompliance, product recalls, and even warning letters.
The historical data from the FDA 483 database shows that CAPA has consistently been the most frequently cited nonconforming sub-element for medical devices each year. Below is a chart based on data from the past three years.
1.2 Regulation Requirements
FDA CAPA Requirements (21 CFR 820.100)
The FDA mandates that medical device companies have documented CAPA procedures to:
• Identify and investigate nonconformances.
• Determine and implement corrective or preventive actions.
• Track and verify the effectiveness of those actions.
ISO 13485:2016 and EU MDR (2017/745)
For companies operating globally, ISO 13485 emphasizes CAPA as a continuous improvement tool within the QMS. Meanwhile, EU MDR integrates CAPA with post-market surveillance (PMS), requiring manufacturers to:
• Use trend analysis to identify CAPA triggers.
• Ensure CAPA actions feed into risk management and design updates.
• Document and maintain CAPA records for regulatory review.
Other Regulatory Agencies
Health Canada, MHRA (UK), and TGA (Australia) also focus on CAPA effectiveness, especially its role in risk-based decision-making and product safety.
2. Common CAPA Challenges (And How to Fix Them)
Even though CAPA is a regulatory must-have, many organizations struggle with execution. Here are some common pitfalls—and how to overcome them.
2.1 Poor Root Cause Analysis: Stopping at Symptoms Instead of Causes
One of the biggest CAPA mistakes is treating symptoms instead of identifying true root causes.
Example:
🔹 A company receives multiple complaints about battery failure in their medical device. The quick fix? Replace the defective batteries. The real issue? A design flaw in the power management system.
How to fix it:
✅ Use structured root cause analysis tools like:
• 5 Whys Analysis – Ask “Why?” multiple times to uncover deeper causes.
• Fishbone Diagram (Ishikawa Diagram) – Visually map out potential causes.
• Fault Tree Analysis (FTA) – Identify failure pathways and systemic issues.
2.2 Inadequate Impact Analysis: Failing to Look at the Bigger Picture
Another common mistake is not considering the broader impact of an issue across different product lines, batches, or markets.
Example:
🔹 A device failure is reported in Europe, but the company only investigates the affected batch. They fail to check whether the issue exists in other regions—until a recall is required in the U.S. six months later.
How to fix it:
✅ Perform a retrospective review: Look at other batches, processes, and suppliers to see if similar risks exist elsewhere.
✅ Conduct impact assessments to determine potential patient safety risks.
2.3 Weak Verification of Effectiveness (VoE): No Proof That the Fix Works
Many companies implement CAPAs but fail to verify their long-term effectiveness.
How to fix it:
✅ Clearly define:
• Methodology – How will success be measured?
• Criteria – What defines an effective CAPA?
• Duration & Frequency – How long will verification last? What's the interval of verification?
• Independence – Who will conduct the verification?
2.4 Incomplete Documentation: A Regulator’s Red Flag
We all heard of this: "If it’s not documented, it didn’t happen"— at least in the eyes of auditors.
No matter how carefully we plan or implement the CAPA, if it's adequately documented, it's insufficient.
How to fix it:
✅ Maintain clear, traceable CAPA records, including:
• Investigation reports.
• Risk assessments.
• Verification of effectiveness.
3. Integrating CAPA into the QMS for a Robust Quality System
A well-structured CAPA system should be interconnected with other QMS processes:
3.1 CAPA & Nonconformance Management
Linking CAPA actions to nonconformity investigations ensures systemic improvements.
3.2 CAPA & Internal Audits
Audits should trigger CAPAs for recurring findings, fostering continuous improvement.
3.3 CAPA & Supplier Quality Management
Extending CAPA principles to suppliers to improve overall product quality.
3.4 CAPA & Post-Market Surveillance (PMS)
Ensuring PMS findings (e.g., customer complaints, field failures) feed into CAPA to prevent recurrence.
3.5 CAPA & Data Analysis
Use trend analysis from customer complaints, production data, and PMS to identify issues before they escalate.
Implement AI-driven CAPA tracking software to detect recurring patterns.
3.6 The Feedback Loop
CAPA outcomes should be reviewed in Management Reviews, ensuring leadership oversight and strategic action.
4. Best Practices for an Effective CAPA System
To enhance CAPA effectiveness, organizations should:
✅ Assign clear ownership – Make sure CAPA responsibilities are well-defined.
✅ Use structured tools – 5 Whys, Fishbone Diagrams, Fault Tree Analysis all help dig deeper into root causes.
✅ Ensure effectiveness checks – Don’t just close CAPAs—monitor them over time.
✅ Invest in digital tools – A cloud-based CAPA tracking system improves efficiency and transparency.
✅ Review CAPA trends regularly – Use data to drive preventive actions, not just corrective ones.
Conclusion
When done right, CAPA can be a powerful driver of quality improvement and risk reduction. Organizations that embrace CAPA as a strategic tool—not just a regulatory requirement—see real benefits:
✅ Fewer regulatory findings (FDA 483s, Warning Letters).
✅ Lower product failure rates and recalls.
✅ Improved efficiency and cost savings.
If you’re ready to improve your CAPA system, start by evaluating how well your current process prevents recurring issues. Does it truly drive improvement, or is it just about closing reports for audits?
The companies that get this right don’t just pass inspections—they build safer, more reliable medical devices and gain a competitive edge.
References:
21 CFR Part 820 Quality System Regulation
ISO 14971:2019 Medical devices — Application of risk management to medical devices
Published on: Mar. 18, 2025